
 SILICA COLUMN
SILICA COLUMN
How to set-up a flash chromatography silica column and actually succeed at separation
Overview.
The following content provides a detailed, low tech classical way to run flash chromatography on a silica column. The material presented below represents the collective wisdom generated by many people during a long time. Not once, detailed descriptions similar to this one were published in various books, and still we find ourselves constantly explaining how to set up a column and actually succeed. Even if one has done a few columns before, just take a look at what kind of results can be achieved on a simple column by following the optimal procedure:
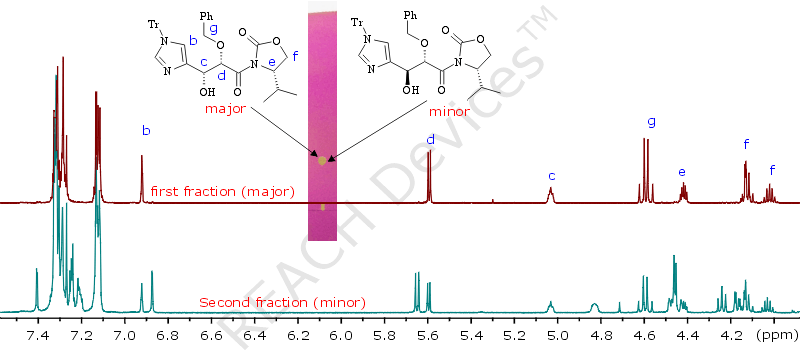
Once upon a time, it was very necessary to separate two diasteroisomers formed in a 7 : 3 ratio. A TLC of their mixture looked hopeless - one spot. And yet, a simple column was set up, according to the guidelines below, and we were able to isolate 300mg the major diastereomer in about 70% recovery. (Luckily it happened to be the higher Rf diastereomer). Picture on the left shows the 1H NMR of fractions obtained.
After that, troublesome separations of alpha-methyl-L-arabinofuranoside (Rf=0.25) from beta-methyl-L-arabinofuranoside (Rf=0.28) on multi-gram scale became simply a routine.
The following procedure for flash chromatography can be summarized in several steps:
- Selection of the elutant to be used
- Selection of silica and apparatus to be used
- Selection of pressure and flow rate
- Running the column and sample collection
What elutant to use?
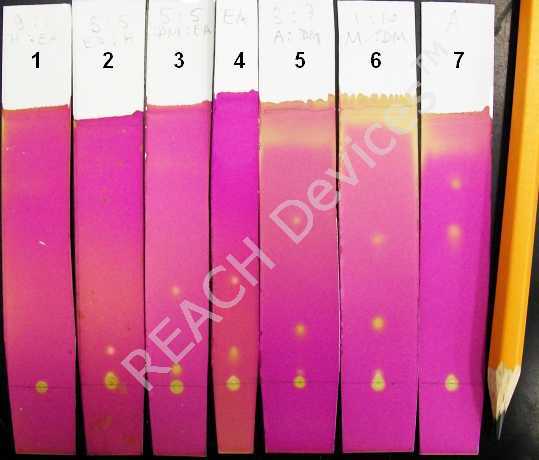
These kind of columns are typically run isocraticly (one solvent for entire separation) or by step-elution (abrupt solvent change). To achieve a reasonable separation and avoid wasting enormous volumes of solvents, the appropriate elutant system(s) should be found first. For this, a series of TLC on silica coated plates must be run prior to the actual column. For a silica column, the elutant 'strength' can be approximately represented by the following sequence (in order of increasing strength):
Perfluoroalkanes (weakest), Hexanes, Pentane, Carbon tetrachloride, Benzene/Toluene, Dichloromethane, Diethyl ether, Ethylacetate, Acetonitrile, Acetone, 2-Propanol/n-Butanol, Water, Methanol, Triethylamine, Acetic acid, Formic acid (strongest)
The rule of thumb is to find such an elutant (or mixture of elutants) in which the spot of the target compound travels about 1/3 of the total elutant path. Or, to be more precise, Rf value should be between 0.25 and 0.35. On the picture to the right the same reaction mixture was subjected to several TLC analyses where the following elutants were used:
| Elutant | Separation | |
| 1 | Hexanes:Ethylacetate 9:1 | Way too weak, nothing moves |
| 2 | Hexanes:Ethylacetate 7:3 | Still too weak |
| 3 | Dichloromethane:Ethylacetate 1:1 | Good for top spot only |
| 4 | Ethylacetate | Also good for the top spot |
| 5 | Dichloromethane:Acetone 7:3 | Tolerable for the bottom spot |
| 6 | Dichloromethane:Methanol 9:1 | Tolerable for the bottom spot |
| 7 | Acetone | Not good for either spot - elutant too strong |
So in this example, if only the top spot is required then elutant 3 or 4 would do the job; if only the following spot is needed then elutant 5 or 6 would do. If BOTH spots are needed there are two choices: use 3 or 4 and continue to elute until all the second fraction will comes out (may be very long and wasteful, up to 100 column volumes). The better choice is to change elutant to 5 or 6 after all the top spot containing fraction comes out. This requires knowledge of the moment when all the top spot compound is out of the column. The best is to use a multi-channel chromatography detector; but it is also possible to do batch TLC before the solvent switch.
Note that the very bottom spot did not move even in straight acetone. If this is the compound of interest, then the silica column separation might be very difficult. Normally, silica is the sorbent of choice for relatively non-polar compounds. For more polar water soluble compounds, reverse phase or other sorbents generally are more suitable. However, the problem with these sorbents is that all good quality reverse phases are MUCH more expensive than silica (10x to 100x). All other sorbents are greatly inferior to silica in resolving power. This is why people persistently attempt to separate awfully polar compounds on silica while using unusual elutant systems. The table below shows a few very interesting examples of such systems:
 |  |  |
| n-Butanol:Acetic Acid:Water 70:7:10(vol) Rf=0.19 | 2-methyl-1-propanol:Acetic Acid:Water 3:1:1(vol) or Acetone:Water:Acetic Acid:Formic Acid 70:20:8:2 and then Phenol:water:Formic Acid 75:24:1. Look at the end of TLC page for actual pictures of such separation! | 1-Propanol:NH4OH(aq, 28%):Water 11:8:2(vol), Rf=0.45 |
| Qi, K.; et al J. Am. Chem. Soc. 2004, 126, 6599. | Detterbeck, F. J. et al Progr. Thin-Layer Chromatogr. Relat. Methods 1971, 2, 235-249. CAS:1972:444944 | Fischer, B. et al J. Med. Chem. 1993, 36, 3937.; Ludwig, J. N.; Eckstein, F. J. Org. Chem. 1989, 54, 631. |
Diffcult cases - Choosing the best elutant mixture

There is always more than one combination of solvents could be used to achieve the desired Rf value (as was outlined above). Which one to use? Well, if the separation looks simple (spots are far apart) then the cheapest non-halogenated solvents should be used. Typically this is hexanes:ethylacetate mixture. Note: cheap hexanes or petroleum ether (especially from plastic storage containers) are always contaminated with non-volatile saturated hydrocarbons. These admixtures will come out on 1H NMR as intensive ugly peaks in 0.9 to 1.5ppm range. This often severely interferes with NMR interpretation. Distil them prior to use or buy a better grade.
Pentane:diethyl ether mixture is very easy to evaporate and so this mixture is highly recommended for unstable or potentially volatile compounds. Also, this mixture allows for complete solvent removal whereas residual traces of hexanes or ethylacetate are almost always seen on 1H NMR if non-crystalline products are obtained after a column.
There are more difficult cases, when spots are overlapping on TLC. Often they can still be resolved by trying dramatically different solvent pairs. Picture to the right shows three TLCs of the same reaction mixture containing benzylated a-hydroxyaldehydes which were eluted in the following mixtures:
| 1. | Hexane:Ethylacetate | 6:4 |
| 2. | Dichloromethane:Ethylacetate | 9:1 |
| 3. | Toluene:Ethylacetate | 8:2 |
Clearly, the mixture 1 is nearly useless for separation; mixture 3 will preclude the use of UV detection (toluene is opaque to UV < 280nm). Mixture 2 seems to be the best for this case.
How much silica should be used?

During the previous step, it should become apparent how difficult the separation is going to be. The closer the spots are, the larger the [silica weight] : [loaded compound weight] ratio will be. The amount of silica needed depends on the silica grain size and grain uniformity. Flash chromatography implies the need to apply some loading-side pressure to the elutant as opposed to older style gravity chromatography. Thus, typical grain size for flash chromatography is smaller and lies in range of 20um to 75um.
| Case | Silica(g) / Compound(g) for 40-75um silica | Silica(g) / Compound(g) for 20-45um silica | ||
| For the top spot | For the following spot | For the top spot | For the following spot | |
| 1 easy separation | 20 : 1 | Too low Rf! | 20 : 1 | Too low Rf! |
| 2 easy separation | 50 : 1 | 50:1 | 20 : 1 | 20 : 1 |
| 3 difficult separation | 100 : 1 | 400 : 1 | 50 : 1 | 100 : 1 |
| 4 troubleshooting needed | 20 : 1, low yield | impractical | 20 : 1, low yield | impractical |
Grain uniformity is the major parameter of any column packing material and has a profound impact on the separation quality. Our lab found a Premium Rf silica from not too popular Sorbent Technologies that is noticeably better than silica from several major suppliers, especially for difficult separations. This statement is from personal observation and as such is subjective. However, it is worthwhile to try a different silica brand if the separation persistently fails.
When calculating the amount of silica to be used, it is much better to measure silica by weight, not by volume, because the density of silica is variable. Silica / compound ratio is usually calculated by relying on the estimated weight of the most abundant component in the mixture. So if there is a mixture composed from 2g of A and 3g of B and the desired amount of silica is 50 : 1 then it is sufficient to use 3 x 50 = 150g silica as opposed to (2 + 3) x 50 = 250g silica.
| Why there is a tail on TLC? | Possible solution |
| Poor solubility | Change elutant - say replace hexane by toluene or dichloromethane. Try crystallization instead of a column |
| Compound is too acidic or sensitive to bases | Add Acetic Acid to elutant. 0.1% to 5% as Rf permits |
| Compound is too basic or sensitive to acids | Add Triethylamine to elutant. 0.05% to 10% as Rf permits. Consider to run an alumina column instead of silica one |
| Compound is moisture sensitive | Try dry neutral Alumina instead of silica. Dry silica under vacuum at 60 - 80C overnight. Increase flow rate and/or decrease amount of silica |
| Compound is too polar for a silica column | Try reverse phase, ion exchange, cellulose, polyamide sorbents. Alumina or Florisyl usually won't work in this case |
Yet another thing to consider before doing a column is the TLC spot shape. Round or slightly elongated spot shapes (cases 1, 2 and 3) usually mean that the column will be successful. A long tail like in case 4 is a sure indication of trouble.
Note that there is no gain in using more silica than needed. Besides wasting time, expensive silica and organic solvents, you will be faced with yet another problem: low recovery. A fraction of the compound applied (2 - 10%) binds to ''active sites'' always present on silica surface and hardly can be eluted from the column packing. This loss will be more prominent when more silica is around or if a compound slowly decomposes/reacts with silica surface.
The importance of the flow rate
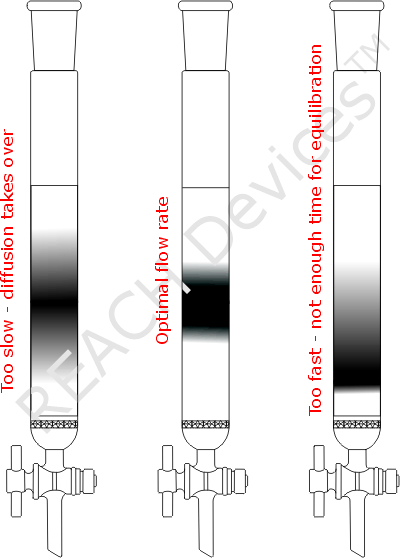
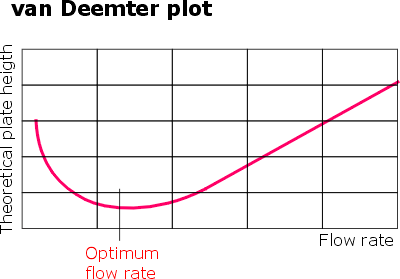
It is widely accepted that elutant/sorbent interaction can be usefully approximated by the van Deemter equation. The common graphic representation of van Deemter equation is shown. A minimum on the graph represents the shortest height of a theoretical plate. This means that a given column reaches the maximum counts of plates which is the best possible separating power. In practice all that translates into a simple fact that for every chromatography separation there is an optimum flow rate value.
The picture of the columns represents the practical outcome when only one compound was loaded onto the column. The same amount of elutant was pushed through all three columns, but flow rates were different. If flow rate was too slow then the substance band diffuses excessively up and down. For overly fast flow rate, there was not enough time for the compound to equilibrate with the column packing. The compound was forcefully dragged down the column leaving behind a long tail.
There are two more implications that result from the van Deemter equation:
- It is not the flow rate, but the linear velocity of the elutant that is important. Hence, small diameter columns must be run much slower that larger diameter ones to achieve the same separation efficiency.
- Finer sorbent particles allow for a longer actual path for the elutant. Thus, the finer the sorbent used the larger the optimum flow rate will be.
So what is better: long and thin columns or short and thick ones? Theoretically, the two are nearly the same, provided the loaded compound mass to sorbent mass ratio is constant. In practice, one should strive for a reasonable [packing length] : [column diameter] ratio. Ratios in range 4:1 to 15:1 work well for nearly all separations. Too long of a packing produces excessive backpressure; too large of a diameter makes it difficult to uniformly apply the mixture onto the column and maintain the packing homogeneity.
Running a column
It is much easier to use an appropriate chromatography detector to run a column, however, most columns are persistantly done without any detection, by blind sample collection, as outlined below.
To get good separation, there is an absolute requirement: after the substance is loaded onto the column, one should not interrupt the flow for more than 2-5 min. Moreover, only one or two gaps like that are allowed. So, no lunch / cigarette breaks or other interruptions. Stopped flow (more than a minute) when the compound is inside the column spells death to the column efficiency.
There are only a few types of inexpensive flash chromatography columns out there on the market. The main differences are if the solvent reservoir is detachable and whether a glass frit is present. The ones with detachable reservoirs are definitely more convenient for larger columns (100g of silica and up). They come fitted with ball joints (as on the picture) or with regular 24/40 joints supplemented by locking nuts. It is important to realize that whenever a detachable reservoir is used, an appropriate locking mechanism must be engaged during the run. A rotovap plastic clip is not adequate here. For smaller runs it is often more convenient to use a single-piece column.
The embedded frit is a mixed blessing. Surely it is quicker to set up a column with a frit. However, poorly made columns have very large dead volumes which do impact separation negatively. Also these frits can clog up, especially with mediocre quality silica. As an alternative, it is possible to use a column without a frit. In these, is a cotton ball plugged in the bottom of the column. The most irritating feature of the cotton ball comes about when a strand of cotton (or glass wool) gets jammed in the teflon stopcock. After that, the stopcock will leak permanently due to a deformed teflon core. Also, some laboratories will choose to cover the cotton ball with a layer of sea sand to provide more uniform flow. While the usefulness of this step is questionable, care must be taken so that the sand does not impact the procedure negatively by mixing with the silica. Therefore, once silica is added, care must be taken to keep the interface between the silica and sand as undisturbed and unmixed as possible.
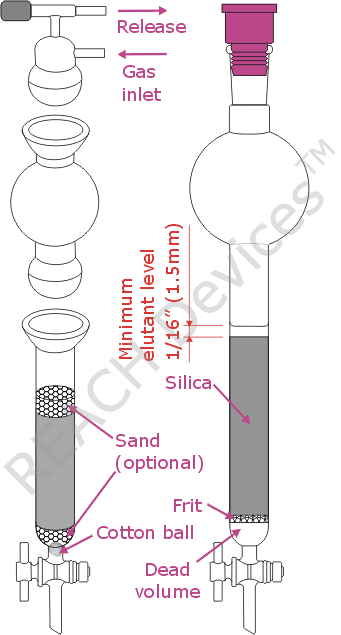
Another thing that must be done is the saturation of the frit or cotton[/sand] with elutant before the introduction of silica. This will remove all air bubbles from said media, improving separation power. Simply add about 5 - 10 mL of elutant to the column, open stopcock intermittently until elutants starts to drip out. No air bubbles should be seen in the column. Drain an excess of elutant, if any.
At this point, the vessel is selected and prepared. Now, the silica must be prepared for loading. First, elutant must be measured off to be used for the rest of the procedure. Usually, the volume needed for a standard chromatography experiment is at least 10x the weight of loaded silica in grams (for a column that will use 100g of silica, 100 x 10 =1000ml). Set this measured elutant aside in a container; if, at any step after this but before the loading of the material to be separated, you need to pour off elutant, return it to the main container for re-use rather than throwing it away.
After preparing the elutant, all silica must now be saturated with it before loading. There are two possible scenarios for this step, depending on whether the separation is easy or difficult (look at the table Suggested silica to compound ratio above).
- Easy: Some elutant is added to the silica, mixing as needed, until the mixture becomes a free-flowing slurry (like a clay suspension).
- Difficult: Silica preequilibration is required. To do this, all measured elutant is added to the silica, and gently stirred for 5-10 min. The silica slurry is then allowed to sediment, and clear layer of elutant is decanted into a separate flask.
In either case, when stirring, avoid magnetic stirrers - they grind the silica particles, and fine dust clogs the column. Use a glass rod or a shaker instead. After that, the silica slurry is transferred to the column. Do not disturb sand on the column bottom (if used). Also, if while silica was being prepared, the sand/cotton ball/frit dried out, make sure to re-saturate them with elutant. After loading, use gentle pressure of argon, nitrogen or compressed air to drain most of the elutant collecting above the slilca layer off the column. Do not use vacuum to drain or run the column. At lowered pressure dissolved gasses will form bubbles throughout the packing and separation efficiency will be decreased dramatically. Another way separation power can be compromised is failing to leave a layer of elutant at the top of the column at any time. There should always be at least 1/16" (1.5mm) of elutant over the silica surface. Gas entering packing creates channels in silica, and separation power is ruined.
After loading the silica, the material solution must now be prepared and loaded. Here again, two methods are possible depending on the solubility of the material to be separated (referred to as "material" later on) in the elutant chosen. To quantify solubility of the material, it is needed to check if it is possible to dissolve all the material to be applied onto column (referred to as "material" later on) in a reasonably small amount of elutant. How small? A good range is 100/20 to 100/10 (5 to 10 mL) of elutant for 100g silica. It is not a good idea to use a different solvent here - the separation will be severely compromised. After quantifying solubility, two possibilities for loading methods exist:
- If solubility is good, the material is simply dissolved in the previously mentioned amount of elutant. The solution is then applied uniformly over the silica surface. A long pipette or a syringe with a thick needle is used. Addition must be carried out in such a manner that the silica surface is not disturbed. (Some people pour sea sand over the silica surface to make it sturdier - this is also not too simple of a procedure, since this possibly second sand-silica interface must also be kept uniform and unmixed. A layer of sand on top of silica layer does noticeably decrease the separation power.)
- If the material to be separated is poorly soluble in the chosen elutant, but TLC looks good (no long tail), then so called "dry loading" is possible. To achieve this, one needs about 1/20 to 1/10 of the dry silica (5 to 10 g in this example) to be mixed with a solution of all material in the appropriate solvent which dissolves it completely. This slurry should be gently evaporated on a rotovap which leaves a dry batch of silica saturated with the material to be separated. It should be a free flowing powder. If it is still an oil, add more silica and repeat the procedure. After this mix is prepared, additional elutant is poured into the silica column to get about one inch of elutant above the silica. The powder is then added, making sure to always retain the 1/16" elutant layer (if all elutant getting absorbed by this addition more elutant must be added). The goal is to form bubble-free slurry over column packing without much disturbance. This does require practice.
After material is loaded, the column is drained carefully until elutant level reaches about 1/16'' (1.5mm) over the silica. Fresh elutant is added then so that its level reaches about 3/8'' (10mm) and this is drained again. This step is repeated one or two more times. Finally all elutant is gently added (or as much as fits) and column begins. The flow rate is adjusted by the column's stopcock and gas pressure.
Gas pressure is typically set at around 2 to 4 psi for 20-45um silica and 1 to 2 psi for 40-75um silica. If the elutant is viscous (contains butanol, water or the like) the pressure may be slightly higher. For glass columns, 7 psi is the ABSOLUTE LIMIT. Delivering pressure through an unsecured septum (as shown in the picture) punctured by a syringe needle adds some protection. If overpressurization occurs, the septum simply pops out, relieving the pressure.
Manual fraction collection...
If there is no detector around, then a blind fraction collection is necessary. One common mistake is the irresistible desire to collect hundreds of very small fractions. There is clearly a cut-off limit to how small a fraction can be, which is dictated by how much silica is being used and the Rf of the target compound. Silica grain size also matters. For Rf=0.3 and, say, 100g silica (40-75um), a good blind fraction size is 100/3 = 33mL, often even 100/2=50mL! This may look shocking but there is no benefit from collecting smaller fractions - they will be joined back together anyway.

For finer silica (20-45um), the fraction size may be smaller (about 100/5=20mL for 100g silica). For lower Rf values, the fraction size becomes even larger. So for a compound Rf=0.15, a fraction size of 60ml (per 100g silica) is just fine.
After fractions are collected (it is normal to collect about 15 fractions and then suspend the elutant flow), one or two wide TLC plates is spotted with all fractions collected. It is a very good idea to spot the starting mixture alongside the fractions (Lane S on the picture).
The group TLC presented here shows that fractions 4 and 5 contain the target component. Also, this plate demonstrates a typical situation when one of the mixture's components happens to be unstable on the silica. Lane 2 and 3 have spots on top AND on the very bottom. This happens because some compound partially decomposed during TLC spotting (remember, all fractions have already gone through the column; theoretically they could not have compounds with so low Rf values). In this particular case, the unstable compound is Bu2Sn(OH)I, which is moisture-sensitive, and slowly hydrolyses on silica onto polymeric dibutyltin oxide and HI.
Pitfalls of the detection: dynamic range, detector's saturation and misprepresentation of concentrations of components
If you never used an UV detector for preparative flash chromatography, please read the following section. Otherwise, an attempt of UV detection may cause more trouble than benefits.
As was shown above, for each preparative chromatographic separation, there is an optimal ratio of quantities of the sample, stationary phase, and elutant.
Typically, flash chromatography purification of 1g of sample will require 20g to 100g of silica, and 200 to 1000mL of total elutant volume. If the column is successful, the desired component should be eluted in no more than 1/10 of the total elutant volume. This gives 1g per 20mL to 1g per 100mL (10 ... 50g/L) concentration range. Note that, in the middle of the sample-rich fraction, sample concentration may be even higher. For compounds with molecular weight of 100-500 g/mol, the molar concentration range will be 0.02 to 0.5 mol/L. The matter does not change whether you purify 10 mg of a target compound using 1 g of stationary phase or 10 g of compound using 500 g of stationary phase.
Lets consider a purification of 1g (0.0073 mol) of methyl benzoate containing 10 mol % (0.00073 mol) of benzyl alcohol as an admixture. If 50 g of silica is used for this procedure, the estimated volume of methyl benzoate-containing fraction will be about 50 mL. This gives 0.0073*1000/50 = 0.15M methyl benzoate concentration. Similarly, the concentration of the admixture (benzyl alcohol) would be around 0.015M.
According to Beer-Lambert law: A = E * b * C, where:
- A is the measured absorbance of a sample, expressed in Absorption Units, AU;
- E is molar absorption coefficient (at a particular wavelength λ) of the compound that is analyzed, expressed in L/(mol * cm);
- b is optical path length, which is equal to the distance between the inner faces of the sample cell, expressed in cm;
- C is the concentration of the compound in solution, expressed in mol/L.
For methyl benzoate E = 14400 L/(mol cm) at λ= 242nm, and for benzyl alcohol E=12 L/(mol cm) at λ= 250 nm.
Thus, measured absorbencies at the following optical path lengths would be:
| Optical path b, mm | Absorbance A, AU | |
| Methyl benzoate, 0.15M | Benzyl alcohol, 0.015M | |
| 10 | 2200 | 0.18 |
| 5 | 1100 | 0.09 |
| 2 | 440 | 0.036 |
| 1 | 220 | 0.018 |
| 0.1 | 22 | 0.0018 |
| 0.01 | 2.2 | 0.00018 |
The calculations demonstrate that on average a UV detector with flow cell of optical path more than 0.01 mm will inevitably saturate during detection of the methyl benzoate. From the other side, 0.01 mm pathlength cell would hardly allow for detection of benzyl alcohol admixture. So efficient flash chromatography UV detection often require the measurement at several path lengths simultaneously to achieve this extended dynamic range.
It must be remembered that the UV detector outputs absorption rather than concentration. Therefore, conversion of detector data into concentration requires knowledge of the molar absorption coefficients, which may differ by a factor of 10000 for different components of the mixture. Therefore, it is very easy to draw the wrong conclusion from a chromatogram (which is a UV absorbtion – vs – time plot).
In routine lab work, the following scenario tends to happen over and over: an unimportant minor admixture shows up as a massive peak while a 100-times more concentrated fraction of the desired compound produces only a miserable bump on the chromatogram. The main fraction gets discarded and an admixture gets all the attention. Therefore, it is good practice to collect all effluent from a column, even if no peaks are detected. A group TLC should be done to link the TLC spots to the fractions collected. Do not destroy column packing before a group TLC proves that all the compounds of interest are actually eluted off the column.